Visualizing Sadie Annotation in GenBank Format¶
After generating Annotations using BLAST implemented in Sadie as AIrr or with HMMER, implemented as Renumbering, you can convert the annotations as features in a Genbank formatted file and then visualize them using your favorite gene browser like Genious. This tutorial shows how you can convert your annotations to GenBank. You can also test it out in a Collab Notebook.
Renumberring HMM¶
The renumbering module in Sadie uses HMMER to annotate the antibody sequences.
import pandas as pd
from Bio.Seq import Seq
from Bio.SeqFeature import FeatureLocation, SeqFeature
from Bio.SeqRecord import SeqRecord
from sadie.renumbering import Renumbering
class ProteinSequenceProcessor:
def __init__(
self,
sequence,
seq_name,
molecule_type="protein",
organism="Human",
scheme="imgt",
region_assign="imgt",
run_multiproc=False,
):
self.sequence = sequence
self.scheme = scheme
self.organism = organism
self.name = seq_name
self.type = molecule_type
self.region_assign = region_assign
self.run_multiproc = run_multiproc
self.map_number = {
"FWR1": "v_gene",
"CDR1": "v_gene",
"FWR2": "v_gene",
"CDR2": "v_gene",
"FWR3": "v_gene",
"CDR3": "d_gene",
"FWR4": "j_gene",
"VGene": "v_gene",
"DGene": "d_gene",
"JGene": "j_gene",
}
def process_sequence(self):
renumbering_api = Renumbering(
scheme=self.scheme, region_assign=self.region_assign, run_multiproc=self.run_multiproc
)
# Run sequence and return renumbering table with sequence_id and sequence
numbering_table = renumbering_api.run_single(self.name, self.sequence)
seq = Seq(self.sequence)
record = SeqRecord(id=self.name, seq=seq, name=self.name)
record.annotations["molecule_type"] = self.type
record.annotations["organism"] = numbering_table.hmm_species[0]
for feature in self.map_number.keys():
_feature = self._get_feature_numbering(numbering_table, feature)
if _feature:
record.features.append(_feature)
return record
def _get_start_stop(self, feature, numbering_table):
start = self.sequence.find(numbering_table[f"{feature.lower()}_aa_no_gaps"][0])
end = start + len(numbering_table[f"{feature.lower()}_aa_no_gaps"][0])
return start, end
def _get_feature_numbering(self, numbering_table, feature):
feature_type = feature
if feature in ["VGene", "JGene", "DGene"]:
return None # The numbering scheme is missing these details (will try and get FW and CDR later)
else:
start, end = self._get_start_stop(feature, numbering_table)
qualifier_dict = {"reference": numbering_table.hmm_species[0]}
try:
qualifier_dict["gene"] = [numbering_table[self.map_number[feature_type]][0]]
except KeyError:
qualifier_dict["gene"] = "Missing"
location = FeatureLocation(start, end)
_feature = SeqFeature(location, type=feature_type, qualifiers=qualifier_dict)
return _feature
if __name__ == "__main__":
seg_name = "PG9"
pg9_aa = "QRLVESGGGVVQPGSSLRLSCAASGFDFSRQGMHWVRQAPGQGLEWVAFIKYDGSEKYHADSVWGRLSISRDNSKDTLYLQMNSLRVEDTATYFCVREAGGPDYRNGYNYYDFYDGYYNYHYMDVWGKGTTVTVSS"
processor = ProteinSequenceProcessor(pg9_aa, seg_name)
genbank_record = processor.process_sequence()
print(genbank_record.format("genbank"))
will print out
LOCUS PG9 136 aa UNK 01-JAN-1980
DEFINITION .
ACCESSION PG9
VERSION PG9
KEYWORDS .
SOURCE .
ORGANISM human
.
FEATURES Location/Qualifiers
FWR1 1..24
/reference="human"
/gene="IGHV3-30*02"
CDR1 25..32
/reference="human"
/gene="IGHV3-30*02"
FWR2 33..49
/reference="human"
/gene="IGHV3-30*02"
CDR2 50..57
/reference="human"
/gene="IGHV3-30*02"
FWR3 58..95
/reference="human"
/gene="IGHV3-30*02"
CDR3 96..125
/reference="human"
/gene="Missing"
FWR4 126..136
/reference="human"
/gene="IGHJ6*04"
ORIGIN
1 qrlvesgggv vqpgsslrls caasgfdfsr qgmhwvrqap gqglewvafi kydgsekyha
61 dsvwgrlsis rdnskdtlyl qmnslrvedt atyfcvreag gpdyrngyny ydfydgyyny
121 hymdvwgkgt tvtvss
//
Now we can write the record to file and visualize¶
with open(f'{seg_name}_hmmer.gb',"w") as handle:
SeqIO.write(genbank_record,handle,"genbank")
You can then load the GenBank file and export the visualization:
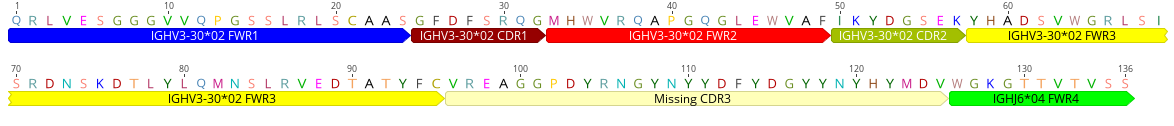
BLAST Annotation Using SADIE¶
Sadie uses igblastn to annotate the sequence provided, which runs through the AIRR API. It can take a single sequence, fasta, or a directory with several files. The output is an AirrTable, which inherits from Pandas DataFrame and has the same functionalities, plus a few more. We parse the AirTable to get the features added to the Genbank annotation.
For AIRR annotation, we have two options. We can annotate the file and convert directly to Genbank format
import pandas as pd
from Bio.Seq import Seq
from Bio.SeqFeature import FeatureLocation, SeqFeature
from Bio.SeqRecord import SeqRecord
from sadie.renumbering import Renumbering
class ProteinSequenceProcessor:
def __init__(
self,
sequence,
seq_name,
molecule_type="protein",
organism="Human",
scheme="imgt",
region_assign="imgt",
run_multiproc=False,
):
self.sequence = sequence
self.scheme = scheme
self.organism = organism
self.name = seq_name
self.type = molecule_type
self.region_assign = region_assign
self.run_multiproc = run_multiproc
self.map_number = {
"FWR1": "v_gene",
"CDR1": "v_gene",
"FWR2": "v_gene",
"CDR2": "v_gene",
"FWR3": "v_gene",
"CDR3": "d_gene",
"FWR4": "j_gene",
"VGene": "v_gene",
"DGene": "d_gene",
"JGene": "j_gene",
}
def process_sequence(self):
renumbering_api = Renumbering(
scheme=self.scheme, region_assign=self.region_assign, run_multiproc=self.run_multiproc
)
# Run sequence and return renumbering table with sequence_id and sequence
numbering_table = renumbering_api.run_single(self.name, self.sequence)
seq = Seq(self.sequence)
record = SeqRecord(id=self.name, seq=seq, name=self.name)
record.annotations["molecule_type"] = self.type
record.annotations["organism"] = numbering_table.hmm_species[0]
for feature in self.map_number.keys():
_feature = self._get_feature_numbering(numbering_table, feature)
if _feature:
record.features.append(_feature)
return record
def _get_start_stop(self, feature, numbering_table):
start = self.sequence.find(numbering_table[f"{feature.lower()}_aa_no_gaps"][0])
end = start + len(numbering_table[f"{feature.lower()}_aa_no_gaps"][0])
return start, end
def _get_feature_numbering(self, numbering_table, feature):
feature_type = feature
if feature in ["VGene", "JGene", "DGene"]:
return None # The numbering scheme is missing these details (will try and get FW and CDR later)
else:
start, end = self._get_start_stop(feature, numbering_table)
qualifier_dict = {"reference": numbering_table.hmm_species[0]}
try:
qualifier_dict["gene"] = [numbering_table[self.map_number[feature_type]][0]]
except KeyError:
qualifier_dict["gene"] = "Missing"
location = FeatureLocation(start, end)
_feature = SeqFeature(location, type=feature_type, qualifiers=qualifier_dict)
return _feature
if __name__ == "__main__":
seg_name = "PG9"
pg9_aa = "QRLVESGGGVVQPGSSLRLSCAASGFDFSRQGMHWVRQAPGQGLEWVAFIKYDGSEKYHADSVWGRLSISRDNSKDTLYLQMNSLRVEDTATYFCVREAGGPDYRNGYNYYDFYDGYYNYHYMDVWGKGTTVTVSS"
processor = ProteinSequenceProcessor(pg9_aa, seg_name)
genbank_record = processor.process_sequence()
print(genbank_record.format("genbank"))

Or fetch a Genbank file from NCBI, then add the features to the file using BioPython. This gives us an exhaustive annotation, which we can visualize as described above.
if __name__ == "__main__":
email = 'example@mail.com'
gene_id = 'GU272045.1'
# Can use a provided sequence
genbank_record = main(email=email, gene_id=gene_id)
with open(f'{gene_id}_complete.gb',"w") as handle:
SeqIO.write(genbank_record,handle,"genbank")
LOCUS GU272045 408 bp mRNA linear PRI 24-JUL-2016
DEFINITION Homo sapiens isolate PG9 anti-HIV immunoglobulin heavy chain
variable region mRNA, partial cds.
ACCESSION GU272045
VERSION GU272045.1
KEYWORDS .
SOURCE Homo sapiens (human)
ORGANISM Homo sapiens
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini;
Catarrhini; Hominidae; Homo.
REFERENCE 1 (bases 1 to 408)
AUTHORS Walker,L.M., Phogat,S.K., Chan-Hui,P.Y., Wagner,D., Phung,P.,
Goss,J.L., Wrin,T., Simek,M.D., Fling,S., Mitcham,J.L.,
Lehrman,J.K., Priddy,F.H., Olsen,O.A., Frey,S.M., Hammond,P.W.,
Kaminsky,S., Zamb,T., Moyle,M., Koff,W.C., Poignard,P. and
Burton,D.R.
CONSRTM Protocol G Principal Investigators
TITLE Broad and potent neutralizing antibodies from an African donor
reveal a new HIV-1 vaccine target
JOURNAL Science 326 (5950), 285-289 (2009)
PUBMED 19729618
REFERENCE 2 (bases 1 to 408)
AUTHORS Chan-Hui,P.-Y.
TITLE Direct Submission
JOURNAL Submitted (04-DEC-2009) In Vitro Pharmacology, Theraclone-Sciences,
1124 Columbia Street, Seattle, WA 98104, USA
FEATURES Location/Qualifiers
source 1..408
/organism="Homo sapiens"
/mol_type="mRNA"
/isolate="PG9"
/db_xref="taxon:9606"
CDS <1..>408
/note="anti-HIV antibody"
/codon_start=1
/product="anti-HIV immunoglobulin heavy chain variable
region"
/protein_id="ADA54566.1"
/translation="QRLVESGGGVVQPGSSLRLSCAASGFDFSRQGMHWVRQAPGQGLE
WVAFIKYDGSEKYHADSVWGRLSISRDNSKDTLYLQMNSLRVEDTATYFCVREAGGPDY
RNGYNYYDFYDGYYNYHYMDVWGKGTTVTVSS"
FWR1 1..72
/gene="IGHV3-33*05"
/reference="human"
CDR1 73..96
/gene="IGHV3-33*05"
/reference="human"
FWR2 97..147
/gene="IGHV3-33*05"
/reference="human"
CDR2 148..171
/gene="IGHV3-33*05"
/reference="human"
FWR3 172..285
/gene="IGHV3-33*05"
/reference="human"
CDR3 286..375
/gene="IGHD3-3*01"
/reference="human"
FWR4 376..408
/gene="IGHJ6*03"
/reference="human"
VGene 1..293
/gene="IGHV3-33*05"
/species="human"
DGene 328..355
/gene="IGHD3-3*01"
/species="human"
JGene 356..408
/gene="IGHJ6*03"
/species="human"
ORIGIN
1 cagcgattag tggagtctgg gggaggcgtg gtccagcctg ggtcgtccct gagactctcc
61 tgtgcagcgt ccggattcga cttcagtaga caaggcatgc actgggtccg ccaggctcca
121 ggccaggggc tggagtgggt ggcatttatt aaatatgatg gaagtgagaa atatcatgct
181 gactccgtat ggggccgact cagcatctcc agagacaatt ccaaggatac gctttatctc
241 caaatgaata gcctgagagt cgaggacacg gctacatatt tttgtgtgag agaggctggt
301 gggcccgact accgtaatgg gtacaactat tacgatttct atgatggtta ttataactac
361 cactatatgg acgtctgggg caaagggacc acggtcaccg tctcgagc
//
and can be visualized as
